
前回の記事では配列データ(Fastqファイル)のQiime2へのインポートを行い,.qzaファイルの生成を行いました。
今回の記事では,Cutadaptパッケージを用いて得られた.qzaファイルの配列情報からプライマー配列を除去していきたいと思います。
目次 非表示

私の解析環境
使用機器:MacBook Pro 14インチ (2023)
チップ:Apple M2 Pro
メモリ:32GB
macOS:Sonoma 14.4.1
Cutadaptによるプライマー配列の除去
今回使用しているテストデータは,MtInsectsプライマーを用いて増幅された配列データ(Fastqファイル)です。
MtInsectsプライマーの配列は以下の通りです。
| プライマー名 | プライマー配列(5′ → 3’) | 塩基数 |
| MtInsects-16S-F | NNN-GGACGAGAAGACCCTWTAGA | 23 |
| MtInsects-16S-R | NNN-ATCCAACATCGAGGTCGCAA | 23 |
以下の手順でプライマー配列を除去します。
1. 前回の記事の続きから開始します。
Qiime2が稼働していることを確認します。稼働していない場合は下記コマンドでアクティベートします。
conda activate qiime2-amplicon-2024.2
2. 作業ディレクトリが01qiime2(/Users/user/Desktop/MtInsects/01qiime2)であることを確認します。
ディレクトリが異なる場合は下記コマンドで移動します。
cd cd /Users/user/Desktop/MtInsects/01qiime2
3. 下記コマンドでCutadaptによるMtInsectsプライマー配列の除去を行います。
qiime cutadapt trim-paired --i-demultiplexed-sequences ./01output/qza/00demux.qza --p-front-f GGACGAGAAGACCCTWTAGA --p-front-r ATCCAACATCGAGGTCGCAA --o-trimmed-sequences ./01output/qza/01primer-trimmed-demux.qza --p-discard-untrimmed true --p-cores 30 --verbose
Terminal画面上に以下の表示がされていればプライマー配列の除去が完了しています。
Terminal画面
Saved SampleData[PairedEndSequencesWithQuality] to: ./01output/qza/01primer-trimmed-demux.qza
4. 下記コマンドでCutadapt前の配列データから可視化用のqzvファイルを作成する。
qiime demux summarize --i-data ./01output/qza/00demux.qza --o-visualization ./01output/qzv/00demux.qzv
5. 下記コマンドでCutadapt後の配列データから可視化用のqzvファイルを作成する。
qiime demux summarize --i-data ./01output/qza/01primer-trimmed-demux.qza --o-visualization ./01output/qzv/01primer-trimmed-demux.qzv
6. Qiime2 Viewで作成した.qzvファイルを可視化します。
ページの上段の「Interactive Quality Plot」タブを選択し,Quality Plotの横軸のSequence Baseの値が減少していることを確認します。
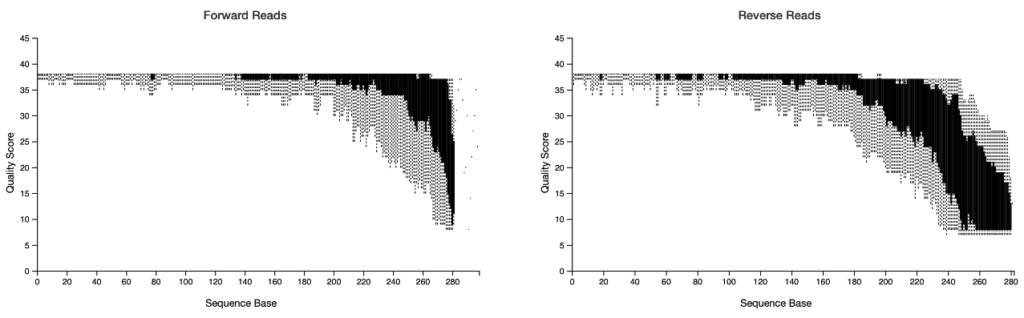
また,ページ下段のDemultiplexed sequence summaryの98%の塩基数(nts)が減少していることを確認します。

このテストデータは2×300 bpでシーケンスされているので,プライマーの塩基数(20塩基)を差し引いて280 bp程度に塩基数が減少していることを確認します。
280 bp程度に減少しているので,テストデータのプライマー配列が除去されていることが確認できました。
最後に
Cutadaptによるプライマー配列の除去方法を解説しました。
次回はDADA2によるキメラ配列除去などのデータ処理を解説します。