
前回の記事では,Cutadaputを使ってプライマー配列の除去を行いました。
今回は,Qiime2内に実装されているDADA2を用いてリード配列のトリミング,ペアエンドリードのマージ,配列クラスタリングによるASVの構築,キメラ配列の除去など行います。
トリミング値の設定方法
DADA2ではトリミング値を設定する必要があります。
初めにトリミング値を設定するために「シーケンスされた配列のクォリティ値」と「オーバーラップ領域」を確認します。
まず,前回の記事と同じように Qiime2 viewで配列データのクォリティを確認します。
前回作成した「demux.qzv」をQiime2 Viewにドラッグ&ドロップし,「Interactive Quality Plot」のタブを開きます。
一般に ,シーケンスされた配列のクオリティ値は後半に向かって低下していきます。ここではクオリティ値が低下する配列位置を確認します。
Plot上で左側から右側に向かってカーソルを移動させると下の表の数値が変化すると思います。
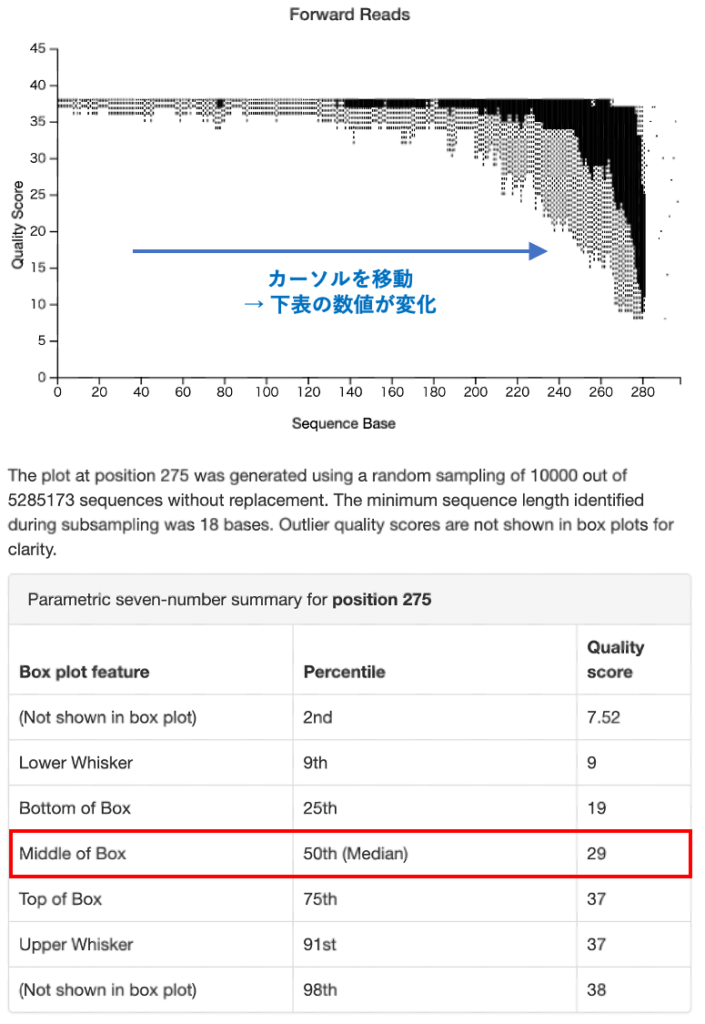
このとき,赤枠内で示した「Middle of Box」の値を参考にします。
この値が30を下回り始めたリード以降は低品質な配列と判断します。
今回のテストデータではforward配列の場合275 bpから,reverse配列は234 bpあたりからクオリティが低下しています。
従って,少なくともForward側は275 bp,Reverse側は234 bp以降の配列は除去するべきだと分かります。
下記図にCutadaptを施した配列データの状況を整理しました。
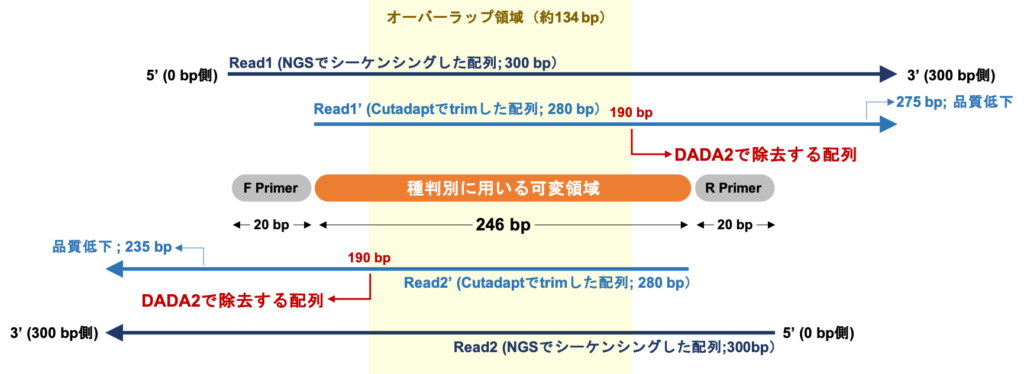
今回のテストデータはMiseqを用いてペアエンド(2×300 bp)でシーケンスされています。
Miseqでは1st PCRのプライマー配列以降の配列からシーケンスされ,Forward側であればForward Primer (F- Primer)を含んで300 bpの生データ(Fastqファイル)が出力されていると思います。
前回の記事では,シーケンスされた300 bpの配列の内,CutadaptでPrimer配列(20 bp)を除去しました。
従って,作成された「01primer-trimmed-demux.qza」には水色で示した配列(280 bp)が収載されています。
オーバーラップ領域とは,Forward側とReverse側からシーケンスした配列が重なる部分を指します。
一般に,オーバーラップ領域は種判別に用いる可変領域上で20 bp以上の塩基数を確保することが望まれます。
MtInsects-16Sの可変領域は246bpです。
今回のテストデータでは両側から300bpシーケンスされているため,種判別に用いる可変領域を通り越して逆側のPrimerやアダプター配列もシーケンスされていることが分かります。
(Forward側からシーケンスした配列の場合,逆側のReverse側のPrimerやアダプター配列までシーケンスが進んでいる状態)
逆側のプライマーやアダプター配列が残っていると正確にマージ配列が作成されないので,DADA2で確実に除去します。
従って,シーケンスした配列のうち190bp以降の配列を除去すれば,逆側のプライマーやアダプター配列は除去され,オーバーラップ領域も134 bp確保できることが分かります。
DADA2を用いた配列データの処理
それでは,以下のコマンドでForward側およびReverse側からシーケンスした配列のうち190bp以降の配列を除去します。
qiime dada2 denoise-paired --i-demultiplexed-seqs ./01output/qza/01primer-trimmed-demux.qza --p-trim-left-f 0 --p-trim-left-r 0 --p-trunc-len-f 190 --p-trunc-len-r 190 --o-representative-sequences ./01output/qza/02rep-seqs-dada2_f190_r190.qza --o-table ./01output/qza/02table-dada2_f190_r190.qza --o-denoising-stats ./01output/qza/02stats-dada2_f190_r190.qza --p-n-threads 0 --verbose
下記コマンドでマージした配列を確認するための.qzvファイルを生成します。
qiime metadata tabulate --m-input-file ./01output/qza/02stats-dada2_f190_r190.qza --o-visualization ./01output/qzv/02stats-dada2_f190_r190.qzv
Qiime2 viewを使って作成した.qzvを可視化します。
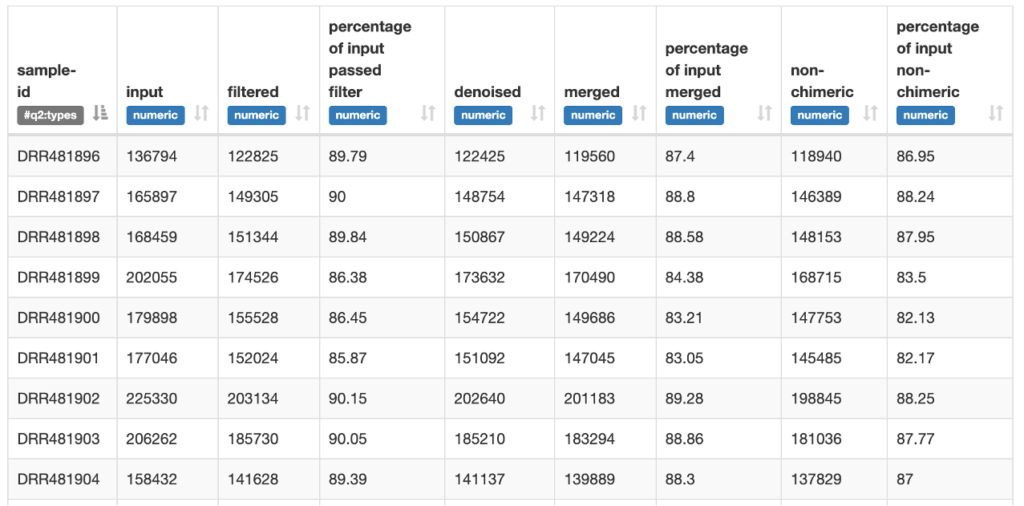
| 項目 | 内容 |
| input | 入力されたリード数 |
| filtered | トリミング後のリード数(今回は190bp以降を除去した後のリード数) |
| denoised | Denoising後のリード数 |
| merged | F-,R-側からシーケンシした配列をマージした後のリード数(オーバーラップが20bp以下だと大幅に減少) |
| Non-chimeric | 最終的に生成された配列のリード数 |
今回の結果では,inputしたリード数がfiltering,denoising,mergingを経由しても極端にリード数が減少しておらず,Non-chimeric配列がinput配列の80%以上を確保できていることが確認できます。
オーバーラップ領域も136 bp程度確保できているので適切にDADA2で処理できたと考えられます。
続いてSingleton, Doubleton, Tripletonを除くため,下記コマンドで4リード未満の配列を削除しておきます。
qiime feature-table filter-features --i-table ./01output/qza/02table-dada2_f190_r190.qza --p-min-frequency 4 --o-filtered-table ./01output/qza/03frequency-filtered-table.qza
フィルタリング後の配列データを作成します。
qiime feature-table filter-seqs --i-data ./01output/qza/02rep-seqs-dada2_f190_r190.qza --i-table ./01output/qza/03frequency-filtered-table.qza --o-filtered-data ./01output/qza/03filtered-rep-seqs.qza
データのエクスポート
DADA2で処理した配列データをtsvファイルに出力していきます。
初めにフィルタリング後の配列データをエクスポートします。配列データは「dna-sequences.fasta」として出力されます。
qiime tools export --input-path ./01output/qza/03filtered-rep-seqs.qza --output-path ./01output/other/
ファイル名を「dna-sequences.fasta」から「Representative_seq.fasta」に変更します。
mv ./01output/other/dna-sequences.fasta ./01output/other/Representative_seq.fasta
下記コマンドでフィルタリング前のテーブルデータをエクスポートする。(フィルタリングできているかの確認するため)
qiime tools export --input-path ./01output/qza/02table-dada2_f190_r190.qza --output-path ./01output/other/
出力されたbiomファイルからtsvファイルを作成する。
biom convert -i ./01output/other/feature-table.biom -o ./01output/other/02feature-table.tsv --to-tsv
同様にフィルタリング後のテーブルデータをエクスポートする。
qiime tools export --input-path ./01output/qza/03frequency-filtered-table.qza --output-path ./01output/other/
biomファイルからtsvファイルを作成する。
biom convert -i ./01output/other/feature-table.biom -o ./01output/other/03feature-filtered-table.tsv --to-tsv
フィルタリング前後のtsvファイルを開いてみましょう。フィルタリング後の行数がフィルタリング前と比較して減少していると思います。
ここまででDADA2を用いた配列データの処理が完了しました。Qiime2をディアクティベートします。
conda deactivate
最後に
DADA2を用いたデータ処理の方法を解説しました。
次回からBlast検索の方法を解説していきます。